Marfan Syndrome
Introduction
Marfan Syndrome is a genetic disorder that affects the connective tissue in the heart, blood vessels, lungs, eyes, bones, and ligaments. It is one of the most common inherited connective tissue disorders and can affect men and women of all backgrounds. (Additional Background)
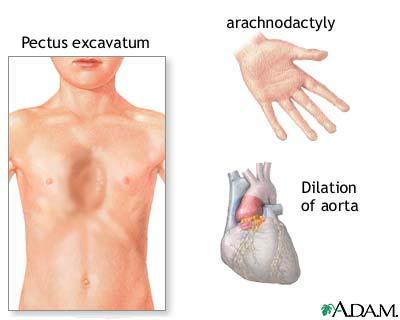 Symptoms
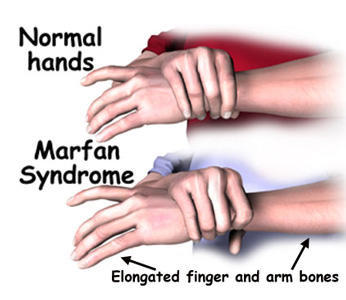
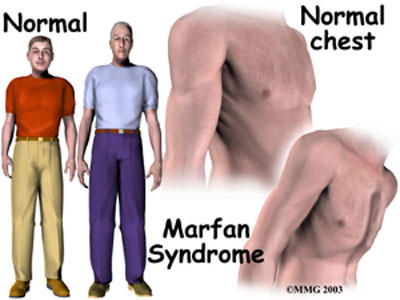
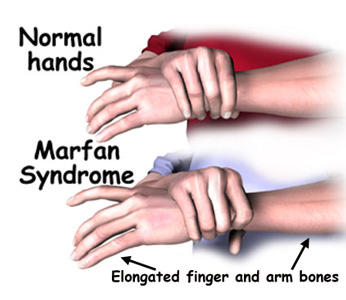
Symptoms The most noticeable symptoms consists of tall slender bodies and appendages. Furthermore, heart problems and other internal problems can occur from the disorder. However, symptoms can be mild or severe in cases. More specific and additional symptoms can be found at NCBI's Online Mendelian Inheritance in Man Website.
The Cause of Marfan Syndrome
Marfan Syndrome has been linked to the protein, fibrillin. Fibrillin is encoded from the FBN1 gene on chromosome 15. The protein is crucial for the formation of elastic fibers that provide structural support in connective tissues. A mutation in the FBN1 gene renders the fibrillin protein nonfunctional and connective tissues can not be properly formed.
Data from several studies suggested fibrillin as a possible candidate gene for Marfan syndrome (154700). Fibrillin was immunolocalized to the ciliary zonule (Sakai et al., 1986), a ligamentous structure consisting mainly of fibrillin microfibrils which is characteristically affected in the Marfan syndrome. Hollister et al. (1990)demonstrated abnormal immunohistochemical patterns in the skin and cultured fibroblasts of patients with the Marfan syndrome. By pulse-chase analysis, Milewicz et al. (1992) demonstrated reproducible patterns of abnormal fibrillin-1 synthesis, secretion, or extracellular matrix utilization in the vast majority of fibroblast cell cultures from Marfan syndrome patients. Furthermore, the Marfan phenotype had been mapped by linkage studies to the same region of chromosome 15 that contained the fibrillin gene. The demonstration of linkage between the fibrillin gene (as recognized by a TaqI restriction site polymorphism) and the Marfan phenotype, combined with the demonstration of point mutations within the fibrillin gene in patients with classic Marfan syndrome (Dietz et al., 1991), concluded the proof that FBN1 is 'the Marfan gene.' In a letter received February 4, 1992, Hayward et al(1992) stated that of the 2 fibrillin mutations discovered up to that time, arg239-to-pro (now designated as codon 1137; 134797.0001) had been found twice in 111 cases and cys1409-to-ser (now designated as codon 2307; 134797.0002) had been found once in 140 cases (mutations being scored once for each sporadic case and once for each family segregating the gene). They concluded that there are likely to be many different FBN1 mutations responsible for the Marfan syndrome.
Navigate through the following link (Marfan Cause) to understand more about the cause of Marfan Syndrome.
Genetic Information
mRNA Reference Sequence - 9749 bp
Protein Reference Sequence - 2871 aa
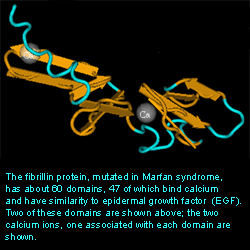 Representative portion of the 3-D structure of fibrillin with two Ca ions in two separate domains.
Representative portion of the 3-D structure of fibrillin with two Ca ions in two separate domains.
courtesy of NCBI
Gene Expression
cDNA sources - NCBI's UniGene
blood, bone, bone marrow, brain, colon, eye, heart, kidney, larynx, liver, lung, lymph node, mammary gland, muscle, ovary, pancreas, peripheral nervous system, placenta, prostate, skin, soft tissue, stomach, testis, thymus, uterus, vascular,embryo, juvenile, adult
Homologs
M. musculus
ref:NP_032019.1 - fibrillin 1 [Mus musculus] 96.17 % / 2871 aa (see ProtEST)
R. norvegicus
ref:NP_114013.1 - fibrillin-1 [Rattus norvegicus] 95.89 % / 2871 aa (see ProtEST)
Treatment
According to the Merck Manual:
There is no cure for Marfan syndrome nor any way to correct the abnormalities in the connective tissue. Treatment is aimed at fixing abnormalities before dangerous complications develop. Some doctors prescribe drugs, such as beta-blockers, that make blood flow more gently through the aorta. However, whether these drugs help is controversial. If the aorta has widened or developed an aneurysm, the affected section can be repaired or replaced surgically. A displaced lens or retina can usually be reattached surgically. Years ago, most people with Marfan syndrome died in their 40s. Now, most people with Marfan syndrome live until their 60s. Prevention of aortic dissection and rupture probably explains why the life span has been lengthened.
Other Resources of the Marfan can be obtain by the links below.
The National Marfan Foundation - This link contains background information, related disorders, supports services, possible grant and research funding, and other significant information
http://www.americanheart.org/presenter.jhtml?identifier=4672 - This link provides more general background about Marfan Syndrome
http://marfan.stanford.edu/ - Information about the Stanford University Marfan Clinic
posted by Anonymous @ 2:34 PM
![]()
![]()

{kind=link}
{kind=link}
{kind=link}
0 Comments:
Post a Comment
Subscribe to Post Comments [Atom]
<< Home