Fibrillin 1
(NP_000129) is encoded from the FBN1 gene on chromosome 15 and is a crucial component of extracellular microfibrils. As a result, Fibrillin 1 is found in the elastic and non elastic connective tissue of the body. Point mutations in the FBN1 gene have been demonstrated to affect the function of Fibrillin 1. This behavior has been well documented as the cause of Marfan Syndrome. The following websites and databases were used to understand more about the structure and function of Fibrillin 1 protein.
Primary/Secondary Structure
The FASTA format of Fibrillin 1 was crucial to most database searches and illustrated the primary structure of Fibrillin 1:
FASTA FORMAT – from NCBI
Compute pI/MWProvided information of the length, molecular weight, and pI
Length – 2871
Molecular Weight – 312237.48
pI – 4.81
COILSProvided prediction of where coil regions are in a protein based on a 14, 21, and 28 window 1frame of an amino acid chain. Result are illustrated several ways (Graphical shown below). Peaks above 50% indicate a strongly probability that coils exist. For Fibrillin 1, very few coil regions were observed.
 Tertiary Structure
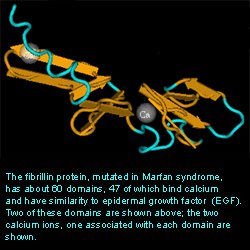
Tertiary StructureInterProScan was useful to relating the Fibrillin 1 protein to the structural domains of other proteins. It provided many follow up websites that included information of the protein family, structural features, and examples of the many domains. One downfall of the program was the long time to provide results. However, the amount of information provided was useful and worth the wait. The results are shown below. As you can see, the matches indicated Ca2+ binding domains which support the function of the protein.
Full Results:
SEARCH RESULTSNCBI’s Structure Database was useful in providing a three dimensional crystal structure of the Fibrillin 1 protein. Only a respresentative portion was shown. The entire protein contains about 60 domains with 47 being able to bind Ca2+.
 Postranslational Modications
Postranslational Modications Postranslational modifications is important to the function of many proteins. Certain websites contain programs that can calculate where certain modifications can take place.
SulfinatorThis website predicted tyrosine sulfation sites.
Fibrillin 1 demonstrated 4 sites out of 93 tyrosines (See Below)
Tyrosine Positions-
434
1004
2849
2853
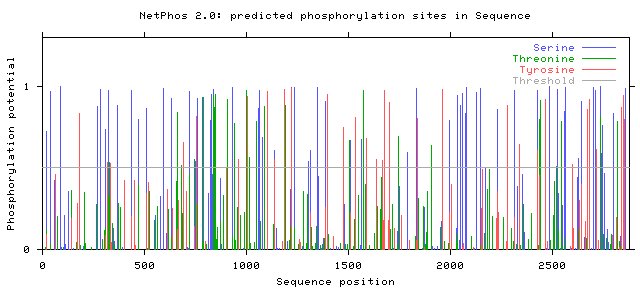
Phosphorylation PredictionThis website predicted the phosphorylation of serine, threonine, and serine. The data can be several formats, including graphical (See Below). Peaks above the threshold indicate that phosphorylation will most likely occur.
Number of Phosphorylation sites predicted in Fibrillin 1: Ser: 65 Thr: 26 Tyr: 32
 Glycosylation Prediction
Glycosylation PredictionThis website predicted O-glycosylation sites in mammalian proteins
Fibrillin 1 illustrated two sites of O-glycosylation
Threonine - 371 and 2101
Protein Localization
TargetPThis site was useful in estimating the location of a protien in the body
Fibrillin was theorized to be localized in
secretory pathway - 86%
mitochondrion - 13%
other - 1%









.0.gif)
 metal binding domain
metal binding domain
.0.gif)












{kind=link}
{kind=link}
{kind=link}
{kind=link}