Transmissible Spongiform Encephalopathies (TSEs)
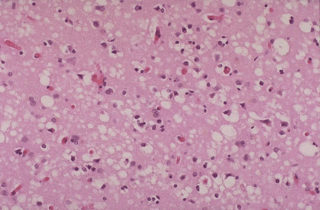 Introduction
IntroductionThe prion diseases are a large group of related neurodegenerative conditions, which affect both animals and humans. Included are Creutzfeldt-Jakob disease (CJD) and Gerstmann-Sträussler-Scheinker (GSS) in humans; bovine spongiform encephalopathy (BSE), or mad cow disease, in cattle; chronic wasting disease (CWD) in mule deer and elk; and scrapie in sheep. These diseases all have long incubation periods but are typically rapidly progressive once clinical symptoms begin. All prion diseases are fatal, with no effective form of treatment currently; however, increased understanding of their pathogenesis has recently led to the promise of effective therapeutic interventions in the near future.
Prion diseases are unique in that they can be inherited, they can occur sporadically, and they can be infectious. The infectious agent in the prion disease is composed mainly or entirely of an abnormal conformation of a host-encoded glycoprotein called the prion protein. The replication of prions involves the recruitment of the normally expressed prion protein, which has mainly an alpha-helical structure, into a disease-specific conformation that is rich in beta-sheet.
The first of these diseases to be described was scrapie, a disease of sheep recognized for over 250 years. This illness, manifested by hyperexcitability, itching, and ataxia, leads to paralysis and death. It is called scrapie because of the tendency of affected animals to rub against the fences of their pens in order to stay upright, reflecting their cerebellar dysfunction. The transmission of this disease was demonstrated first in 1943 when a population of Scottish sheep was accidentally inoculated against a common virus using a formalin extract of lymphoid tissue from an animal with scrapie (Gordon, 1946). Accidental transmission of prions is a recurrent event in the history of these agents and is related to their unusual biophysical properties.
excerpt from eMedicine.com
This hypothesis was initially greeted with great skepticism in the scientific community; now it represents the current dogma, and Prusiner won the 1998 Noble Prize for Science. This hypothesis suggests that prions contain no nucleic acid and are referred to as PrPSc. The latter represents a conformationally modified form of a normal cellular PrPC, which is a normal host protein found on the surface of many cells, in particular neurons. PrPSc, when introduced into normal healthy cells, causes the conversion of PrPC into PrPSc, initiating a self-perpetuating vicious cycle.
excerpt from eMedicine.com
Although Prion diseases can be spontaneous, infectious as well as genetic, the focus here is on the hereditary disease....
• In the US: The most common prion disease is CJD, with a uniform incidence of approximately 1 case per million population both in the United States and internationally. Familial forms of prion diseases, such as GSS and fatal familial insomnia (FFI), are much more rare. About 10% of cases of CJD are familial, with an autosomal dominant pattern of inheritance linked to mutations in the PRNP gene.
Mortality/Morbidity
Prion-related diseases are relentlessly progressive and invariably lead to death.
• The mean duration of sporadic CJD is 8 months.
• nvCJD has a slightly longer course, with a mean duration of 16 months.
• Familial CJD has a mean duration of 26 months, while GSS has the longest course, about 60 months.
*there are no sex/race predominance of the genetic disease
Creutzfeldt-Jakob Disease (CJD)
• 10–15% of the cases of CJD are inherited
• inherited as autosomal dominant
• mutation of the PRNP gene - most common mutations are:
• a change in codon 200 converting glutamic acid (E) at that position to lysine (K)(thus designated "E200K")[link to a table giving the single-letter code for the amino acids]
• a change from aspartic acid (D) at position 178 in the protein to asparagine (D178N) when it is accompanied by a polymorphism in the gene encoding valine at position 129 . (When the polymorphism at codon 129 is Met/Met, the D178N mutation produces Fatal Familial Insomnia instead.)
• a change from valine (V) at position at position 210 to isoleucine (V210I)
• Extracts of autopsied brain tissue from these patients can transmit the disease to:
• apes (whose PRNP gene is probably almost identical to that of humans).
• transgenic mice who have been given a Prnp gene that contains part of the human sequence.
***These results lead to the important realization that prion diseases can only be transmitted to animals that already carry a PRNP gene with a sequence that is at least similar to the one that encoded the PrPSc. In fact, knockout mice with no Prnp genes at all cannot be infected by PrPSc.
Gerstmann-Sträussler-Scheinker disease (GSS)
• caused by the inheritance of a PRNP gene with a mutations encoding most commonly:
• leucine instead of proline at position 102 (P102L) or
• valine instead of alanine at position 117 (A117V)
• like JCD, GSS is also strongly associated with homozygosity for a polymorphism at position 129 (both residues being methionine).
• Brain extracts from patients with GSS can transmit the disease to
• monkeys and apes
• transgenic mice containing a portion of the human PRNP gene.
Fatal Familial Insomnia (FFI)
People with this rare disorder have inherited
• a PRNP gene with asparagine instead of aspartic acid encoded at position 178 (D178N)
• the susceptibility polymorphism of methionine at position 129 of the PRNP gene.
Extracts from autopsied brains of FFI victims can transmit the disease to transgenic mice.
The normal protein
• is called PrPC (for cellular)
• is a glycoprotein normally found at the cell surface inserted in the plasma membrane
• has its secondary structure dominated by alpha helices (probably 3 of them)
• is easily soluble
• is easily digested by proteases
• is encoded by a gene designated (in humans) PRNP located on our chromosome 20.
PrPSc
The abnormal, disease-producing protein
• is called PrPSc (for scrapie)
• has the same amino acid sequence as the normal protein; that is, their primary structures are identical but
• its secondary structure is dominated by beta conformation
• is insoluble in all but the strongest solvents
• is highly resistant to digestion by proteases
• When PrPSc comes in contact with PrPC, it converts the PrPC into more of itself (even in the test tube).
• These molecules bind to each other forming aggregates.
• It is not yet clear if these aggregates are themselves the cause of the cell damage or are simply a side effect of the underlying disease process.
excerpt from CDC.gov webpage
Unigene
Prion diseases are unique in that they can be inherited, they can occur sporadically, and they can be infectious. The infectious agent in the prion disease is composed mainly or entirely of an abnormal conformation of a host-encoded glycoprotein called the prion protein. The replication of prions involves the recruitment of the normally expressed prion protein, which has mainly an alpha-helical structure, into a disease-specific conformation that is rich in beta-sheet.
The first of these diseases to be described was scrapie, a disease of sheep recognized for over 250 years. This illness, manifested by hyperexcitability, itching, and ataxia, leads to paralysis and death. It is called scrapie because of the tendency of affected animals to rub against the fences of their pens in order to stay upright, reflecting their cerebellar dysfunction. The transmission of this disease was demonstrated first in 1943 when a population of Scottish sheep was accidentally inoculated against a common virus using a formalin extract of lymphoid tissue from an animal with scrapie (Gordon, 1946). Accidental transmission of prions is a recurrent event in the history of these agents and is related to their unusual biophysical properties.
excerpt from eMedicine.com
Etiology
Highly divergent hypotheses have been put forward regarding the makeup of the prions, including that they consist of nucleic acid only or protein only, are lacking both protein and nucleic acid, or are a polysaccharide. The most widely accepted hypothesis, first described by Griffith (Griffith, 1967) and more explicitly detailed by Stanley Prusiner, MD, is the protein only hypothesis (Prusiner, 1998). Prusiner introduced the term prion to indicate that scrapie is related to a proteinaceous infectious particle (PrP) (Prusiner, 1982).This hypothesis was initially greeted with great skepticism in the scientific community; now it represents the current dogma, and Prusiner won the 1998 Noble Prize for Science. This hypothesis suggests that prions contain no nucleic acid and are referred to as PrPSc. The latter represents a conformationally modified form of a normal cellular PrPC, which is a normal host protein found on the surface of many cells, in particular neurons. PrPSc, when introduced into normal healthy cells, causes the conversion of PrPC into PrPSc, initiating a self-perpetuating vicious cycle.
excerpt from eMedicine.com
Although Prion diseases can be spontaneous, infectious as well as genetic, the focus here is on the hereditary disease....
Inherited Prion Diseases
Frequency• In the US: The most common prion disease is CJD, with a uniform incidence of approximately 1 case per million population both in the United States and internationally. Familial forms of prion diseases, such as GSS and fatal familial insomnia (FFI), are much more rare. About 10% of cases of CJD are familial, with an autosomal dominant pattern of inheritance linked to mutations in the PRNP gene.
Mortality/Morbidity
Prion-related diseases are relentlessly progressive and invariably lead to death.
• The mean duration of sporadic CJD is 8 months.
• nvCJD has a slightly longer course, with a mean duration of 16 months.
• Familial CJD has a mean duration of 26 months, while GSS has the longest course, about 60 months.
*there are no sex/race predominance of the genetic disease
Creutzfeldt-Jakob Disease (CJD)
• 10–15% of the cases of CJD are inherited
• inherited as autosomal dominant
• mutation of the PRNP gene - most common mutations are:
• a change in codon 200 converting glutamic acid (E) at that position to lysine (K)(thus designated "E200K")[link to a table giving the single-letter code for the amino acids]
• a change from aspartic acid (D) at position 178 in the protein to asparagine (D178N) when it is accompanied by a polymorphism in the gene encoding valine at position 129 . (When the polymorphism at codon 129 is Met/Met, the D178N mutation produces Fatal Familial Insomnia instead.)
• a change from valine (V) at position at position 210 to isoleucine (V210I)
• Extracts of autopsied brain tissue from these patients can transmit the disease to:
• apes (whose PRNP gene is probably almost identical to that of humans).
• transgenic mice who have been given a Prnp gene that contains part of the human sequence.
***These results lead to the important realization that prion diseases can only be transmitted to animals that already carry a PRNP gene with a sequence that is at least similar to the one that encoded the PrPSc. In fact, knockout mice with no Prnp genes at all cannot be infected by PrPSc.
Gerstmann-Sträussler-Scheinker disease (GSS)
• caused by the inheritance of a PRNP gene with a mutations encoding most commonly:
• leucine instead of proline at position 102 (P102L) or
• valine instead of alanine at position 117 (A117V)
• like JCD, GSS is also strongly associated with homozygosity for a polymorphism at position 129 (both residues being methionine).
• Brain extracts from patients with GSS can transmit the disease to
• monkeys and apes
• transgenic mice containing a portion of the human PRNP gene.
Fatal Familial Insomnia (FFI)
People with this rare disorder have inherited
• a PRNP gene with asparagine instead of aspartic acid encoded at position 178 (D178N)
• the susceptibility polymorphism of methionine at position 129 of the PRNP gene.
Extracts from autopsied brains of FFI victims can transmit the disease to transgenic mice.
Characteristics of the normal and mutated prion protein
PrPCThe normal protein
• is called PrPC (for cellular)
• is a glycoprotein normally found at the cell surface inserted in the plasma membrane
• has its secondary structure dominated by alpha helices (probably 3 of them)
• is easily soluble
• is easily digested by proteases
• is encoded by a gene designated (in humans) PRNP located on our chromosome 20.
PrPSc
The abnormal, disease-producing protein
• is called PrPSc (for scrapie)
• has the same amino acid sequence as the normal protein; that is, their primary structures are identical but
• its secondary structure is dominated by beta conformation
• is insoluble in all but the strongest solvents
• is highly resistant to digestion by proteases
• When PrPSc comes in contact with PrPC, it converts the PrPC into more of itself (even in the test tube).
• These molecules bind to each other forming aggregates.
• It is not yet clear if these aggregates are themselves the cause of the cell damage or are simply a side effect of the underlying disease process.
excerpt from CDC.gov webpage
Homologs
Homologene Unigene
Other Related Information
posted by Anonymous @ 11:23 PM
![]()
![]()

0 Comments:
Post a Comment
Subscribe to Post Comments [Atom]
<< Home